|
|
||||||
|
|
|
|
Download | |||
Diamond Version 5 User Manual: Distances and anglesTable of distances and anglesThis article in brief:
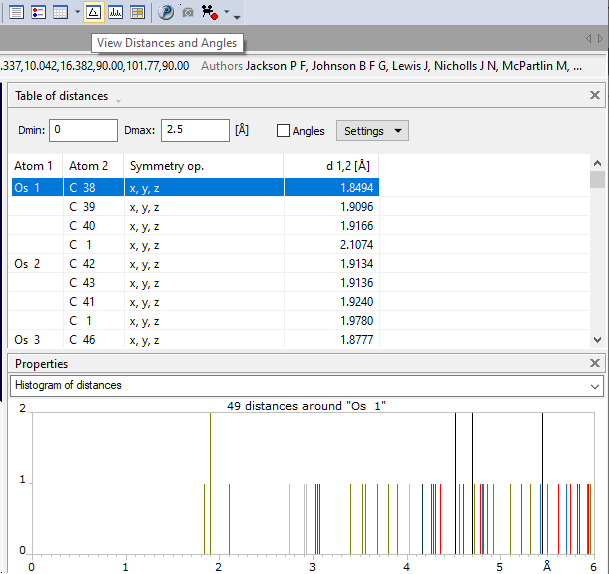
Previous article: Destroying polyhedra Showing the distances and angles tableLike the data sheet or the several object tables, the table of distances and/or angles is displayed in the secondary pane (data pane) of the structure window. To open the table of distances and/or angles, choose the Distances and Angles command from the View menu. If the secondary pane has not yet been opened, the mouse cursor will change to split mode, that means you can shift the border line between graphics and data pane to adjust the appropriate width for the table of distances and angles. You find the distances and angles table in the upper part of the data pane and a distances histogram in the lower part (that means in the "Properties" pane). In addition to the Distances and Angles command from the View menu, you can also use the corresponding button from the main toolbar. (This is highlighted in the screenshot below.)
When the table is displayed the first time for a structure, the distances (and as far as necessary the angles) will be calculated automatically, using the previous settings for the distance range (the default range is 0 to 2.5 Angstroems), for all atom types of the structure. You can stop the calculation with the Ctrl+Break key combination. This may be necessary, if the preset distance range is rather big (for example up to 5 Angstroems) and the structure has many atoms in the parameter list.
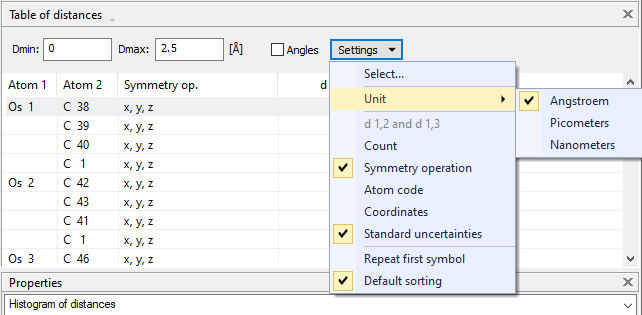
Settings for distances and angles tableAbove the actual table of distances (and angles, if option activated), there is a small dialog bar for the most common settings:
Dmin and Dmax Angles Settings
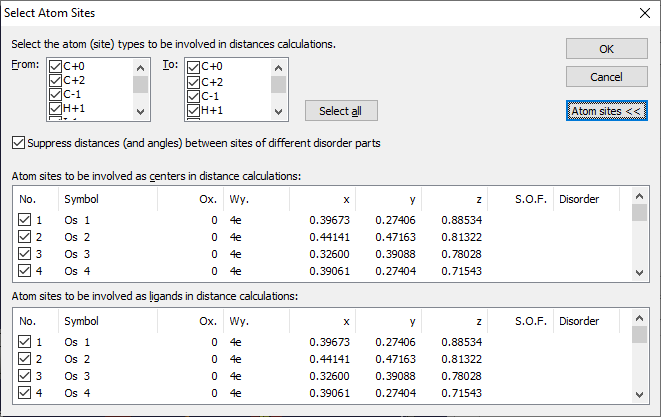
Defining the distance range Define the distance range in the input fields Dmin and Dmax in Angstroems. Enter or change the value(s) and press the Enter or Return key. If you want to have a different unit, use the dropdown menu of the Settings button and choose the appropriate unit from the Unit sub-menu. Selecting which atom sites to regard in calculation By default, all atom sites of the current structure are selected for distance (and angle) calculation, but you can select the atom types (i.e. combinations element/oxidation state) and/or atom sites from the Settings dropdown menu and running the Select... command. This opens the Select Atom Sites dialog in order to select which atom types and atom sites shall be included in the table of distances and angles.
By default, you see only the upper part of the dialog with the atom types. If you want to choose central and/or ligand atoms on atom site level, click on the Atom sites button to open the lower part with the two atom site lists. You can select the sites for central and ligand atoms separately. By default, atoms from all sites are involved (at least as long as the corresponding atom tyes are selected at the top of the dialog). Choose the atom types to be used in distance calculation by setting the corrsponding checkmarks in the lists From and To. If you press the Select all button, atoms of all types will be included both as central atoms and ligands in the distances/angles table. Supressing distances (and angles) between sites of different disorder parts If disorder parts are defined in the atomic parameter list, distances between atom sites belonging to different disorder parts are not calculated and shown in the table of distances and angles (nor in the distances histogram below), provided the check mark is set. If the check mark is not set, the disorder informations will be ignored.
Selecting between distances and angles calculationBy default only distances but no angles are displayed in the table. If angles are displayed yet, clear the checkmark at Angles.
Displaying distances only
Although details of the list format will be treated later, the list displays the following informations, if only distances are displayed. The example calculates distances up to 2.5 Angstroems, collects identical distances and uses standard uncertainties. (The output has been copied directly from the list without reformatting:)
Ti 1 F 8 1.732(4) 3x
F 1 2.026(5) 3x
Ti 2 F 10 1.720(6) 1x
F 6 1.745(4) 1x
F 1 1.895(4) 1x
F 5 1.914(5) 1x
F 3 1.947(4) 1x
F 2 2.037(5) 1x
Ti 3 F 7 1.708(4) 1x
F 9 1.732(4) 1x
F 4 1.874(5) 1x
F 2 1.891(4) 1x
F 3 1.995(5) 1x
F 5 2.002(4) 1x
Ti 4 F 11 1.742(4) 3x
F 4 2.058(4) 3x
F 1 Ti 2 1.895(4) 1x
Ti 1 2.026(5) 1x
F 2 Ti 3 1.891(5) 1x
Ti 2 2.037(5) 1x
F 3 Ti 2 1.947(4) 1x
Ti 3 1.995(5) 1x
F 4 Ti 3 1.874(5) 1x
Ti 4 2.058(5) 1x
F 5 Ti 2 1.914(5) 1x
Ti 3 2.002(5) 1x
F 6 Ti 2 1.745(4) 1x
F 7 Ti 3 1.708(4) 1x
F 8 Ti 1 1.732(4) 1x
F 9 Ti 3 1.732(4) 1x
F 10 Ti 2 1.720(6) 1x
F 11 Ti 4 1.742(4) 1x
Displaying angles only To activate the calculation and display of angles, set the checkmark at Angles. Ensure that the checkmark at d 12 and d 13 is cleared in the dropdown menu from the Settings button. Although only angles will be displayed in this mode, you have to specify a distance range. The reason is that Diamonds first calculates distances in any case, and then calculates angles between all atoms within the (coordination) sphere. (The list has been abbreviated.)
Ti 1 F 8 F 8 100.61(15)
< omitted for brevity >
F 1 Ti 2 Ti 1 147.97(21)
F 2 Ti 3 Ti 2 148.78(25)
F 3 Ti 2 Ti 3 151.05(22)
F 4 Ti 3 Ti 4 143.40(24)
F 5 Ti 2 Ti 3 152.72(22)
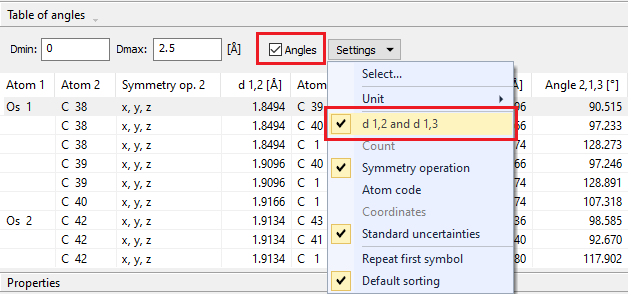
Displaying both distances and angles To see both angles as well as the distances to the two atoms each, ensure that the checkmark is set for both the Angles checkbox and the d 12 and d 13 command in the dropdown menu:
Like for the previous option, Diamond calculates distances first, then angles between all atoms of each (coordination) sphere. For this option, Diamond displays the ligand atoms together with the distances from the central atom. (The list has been abbreviated.)
Ti 1 F 8 1.732(4) F 8 1.732(4) 100.61(15)
< omitted for brevity >
F 1 Ti 2 1.895(4) Ti 1 2.026(5) 147.97(21)
F 2 Ti 3 1.891(5) Ti 2 2.037(5) 148.78(25)
F 3 Ti 2 1.947(4) Ti 3 1.995(5) 151.05(22)
F 4 Ti 3 1.874(5) Ti 4 2.058(5) 143.40(24)
F 5 Ti 2 1.914(5) Ti 3 2.002(5) 152.72(22)
Configuring the format of the table of distances and anglesThe settings menu to configure the distances and angles table is available from both the title bar "Table of distances" or "Table of angles", rsp. above the table as well as from the dropdown menu that opens from the Settings button (see screenshot #2 above). It offers the following settings: Unit: Here you can select the unit for the distance values (Å, pm or nm) in the table of distances and angles. d 1,2 and d 1,3: This option is only available if the Angles checkbox above is selected. For each angle, the individual distances from the central atom(s) to the two ligand atoms is displayed in the table at the top. Count: This option is only available if the Angles checkbox above is not selected. For each central atom, all equivalent distances to ligand atoms with the same distance and the same atom label are counted and given in a single line. In this case, the distance value will be listed only once together with the coordination number, e.g. "6x". This option is deselected automatically if Symmetry op. or Atom code is selected. Symmetry operation: For each ligand atom, the symmetry operation by which it is created from the atomic parameter list is displayed. This option is deselected automatically if Count or Atom code is selected. Atom code: By activating this option, the individual atom codes of the ligand atoms are displayed.
This option is deselected automatically if Count or Symmetry op. is selected. Coordinates: The crystal coordinates of the ligand atoms in the crystal structure (not the coordinates of the corresponding atom in the parameter list!) are displayed if this option is selected. This option is deactivated automatically if Angles is selected. Standard uncertainties: If this option is selected, the standard uncertainties of the distances d 1,2 between central and ligand atoms as well as angles 2,1,3 are displayed. Repeat first symbol: By activating this option, the symbol of the central atom is displayed not only in the line of the first ligand atom but for all ligand atoms. This is useful, if you copy the list, e.g. to a spreadsheet. Normally the central atom is not repeated for each line. Default sorting: This sets (or resets to) the default sort order, which is primarily for the (central) atoms as they appear in the atomic parameter list. You can change the default sort order by clicking on one of the columns in the table, e.g. for increasing or decreasing distance or angle values. A check mark indicates, if the table uses default sort order or not.
The following example compares the usage of coordination numbers (upper part) and symmetry codes in sodium chloride (lower part):
Na1 Cl1 2.810 6x
Cl1 Na1 2.810 6x
Na1 Cl1 x, -0.5+y, -0.5+z 2.810
Cl1 -0.5+x, -1+y, -0.5+z 2.810
Cl1 -0.5+x, y, -0.5+z 2.810
Cl1 -0.5+x, -0.5+y, -1+z 2.810
Cl1 -0.5+x, -0.5+y, z 2.810
Cl1 -1+x, -0.5+y, -0.5+z 2.810
Cl1 Na1 1+x, 0.5+y, 0.5+z 2.810
Na1 0.5+x, y, 0.5+z 2.810
Na1 0.5+x, 1+y, 0.5+z 2.810
Na1 0.5+x, 0.5+y, z 2.810
Na1 0.5+x, 0.5+y, 1+z 2.810
Na1 x, 0.5+y, 0.5+z 2.810
Selecting the central atom from the structure pictureYou can select an atom in the structure picture and run the command Select in Table from the context menu of the structure picture. This automatically selects the corresonding central atom (i.e. atom from the parameter list) in the table of distances (or angles, rsp.). Read more about selections between structure picture and several data tables in the article "Tables of objects like atoms, bonds, molecules etc.".
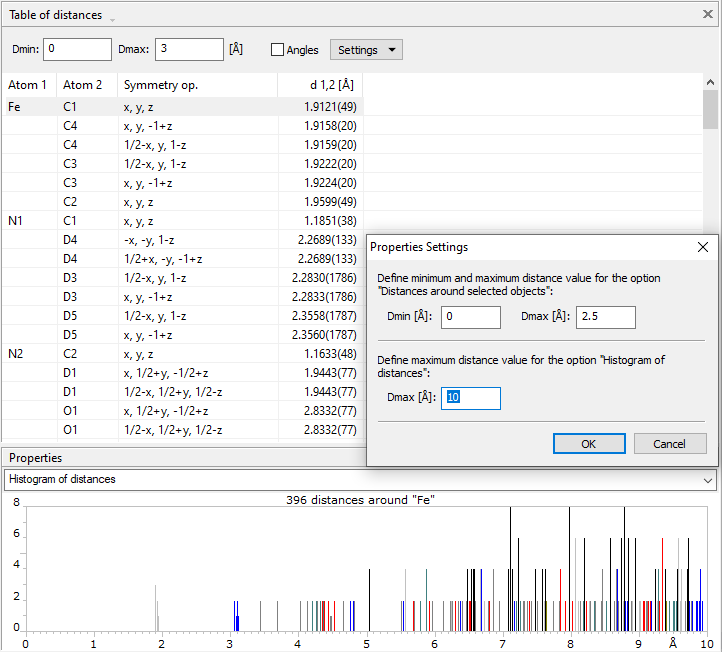
Distances histogram in the Properties paneIn the Properties pane, that means usually below the distances and angles table, a histogram of all distances for the central atom which is currently selected in the table above is displayed. In this histogram, every ligand atom type is represented by a different color, so you can easily distinguish the contribution of the individual atom type ligands to the overall distance histogram. The distance range in the histogram can be changed in the Properties Settings dialog, available through the corresponding command in the context menu of the histogram.
Table of distances up to 3 Å with distance histogram up to 10 Å for central atom "Fe". Data taken from PCD: 1503073 [2].
Previous article: Destroying polyhedra
[1] COD: 9000684. Hazen, R. M.; Finger, L. W.; "Crystal structure and compressibility of zircon at high pressure crystal No. 1, 1 atm - before P"; American Mineralogist, 64, 196-201 (1979) |
|
Page last modified October 12, 2023. Copyright © 2023 Crystal Impact GbR. All rights reserved. Contact Webmaster |