|
|
||||||
|
|
|
|
Download | |||
Diamond Version 5 User Manual: Building up structural partsUsing the Mouse WheelSeveral options to use the mouse wheel to change properties (enlargement factor, zoom in/out, change atom radii) or to change surroundings (blow up polyhedra, expand molecule cluster, etc.)This article describes how to use the mouse wheel to:
Previous article: Pump Up or Shrink Polymeric Frameworks
The so-called "Mouse wheeling" modes are available through the Mouse Wheel Action
sub-menu in the Tools menu. These modes have in common that a tick of the mouse
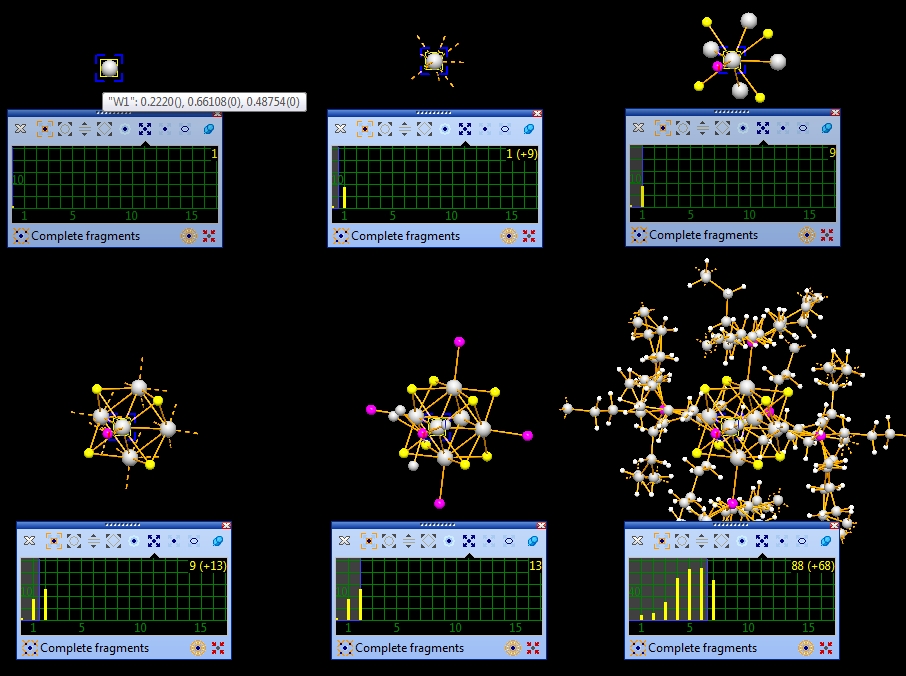
wheel changes a property each. The modes and the properties changed are the following: The wheeling modes "Pump up or shrink", "Expand via contacts", and "Molecule cluster" are demonstrated below. The wheeling mode "Expand selection" is described in the article "Selecting objects". The wheeling modes "Atom radii" and "Bond radii" are described in the articles "Designing atoms" and "Designing bonds and contacts". The usage of the mouse wheel to change the enlargement factor or to zoom in or out is described in the article "Grab mode". Pump up or shrinkThe "Pump up or shrink" wheeling mode adds/removes atoms of the n-th sphere around selected atom, while stepping via broken-off bonds. As an example we use the document COD-10004001.diamdoc: We start with a single atom of the parameter list, that is the first one: "W1", and complete the molecule step-by-step then expand via contacts and H-bonds to neighbouring molecules' atoms and continue with step-by-step building up...
(1) Destroy all ("Build/Destroy/All" or Shift+Ctrl+D). This deletes the whole molecule,
the picture is now blank.
This is the mentioned sequence for "W1" as center. (We omitted some
steps between
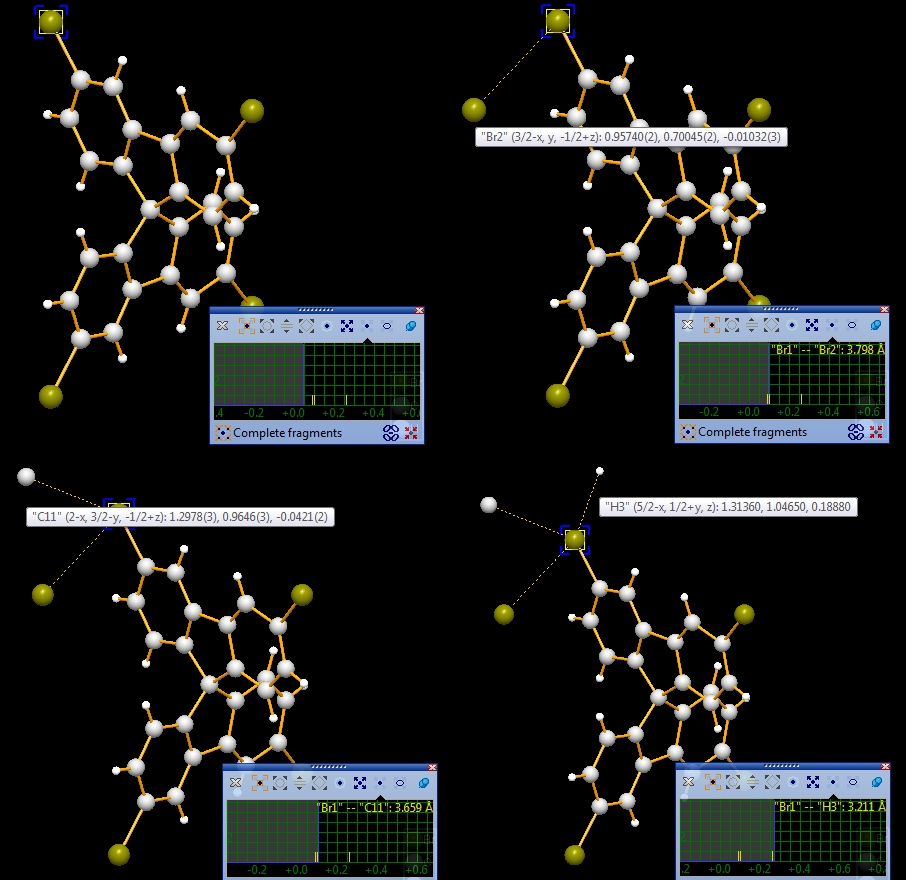
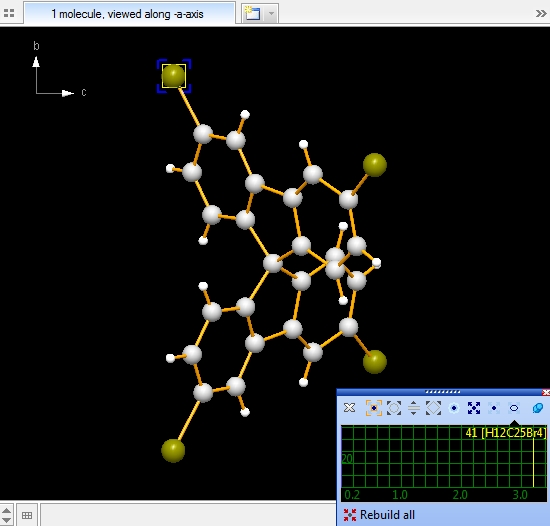
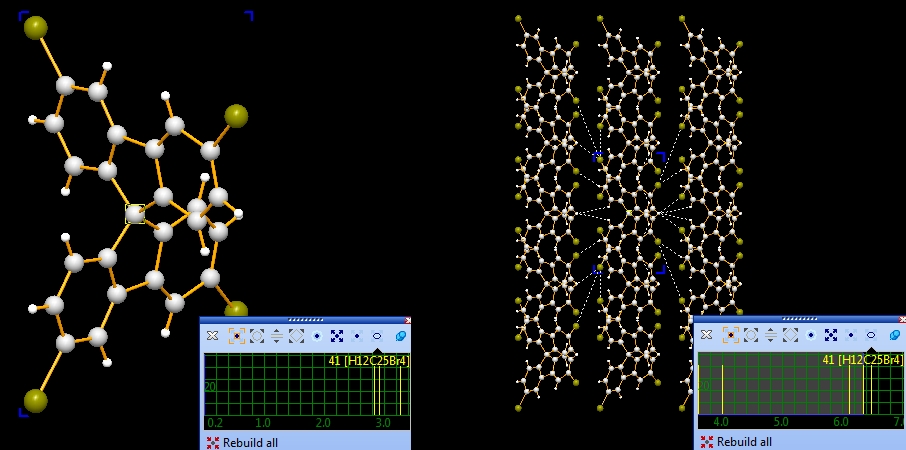
the fifth and the sixth picture): Note: Screenshots have been made with version 4 of Diamond. Expand via contactsIn the following example, we first examine a contact sphere, then try out to find subsequently neighbouring molecules via these contacts. We open the example with one molecule of COD:1501635 from COD-1501635-molecule.diamdoc, click on the "Br1" atom at (x,y,z) (in the top left when viewed along -a-axis), and run the command "Tools"/"Mouse Wheel Action"/"Expand via Contacts". The blue blinking marker appears and the mouse wheeling control window opens for "Expand via contacts" mode. There are three atoms in the neighbourhood of "Br1", not more than the sum of van der Waals radii plus an offset of 0.1 Angstroem away. You get these three neighbours ("Br2", "C11", "H3") by turning a mouse wheel tick each, as the sequence of four pictures show below:
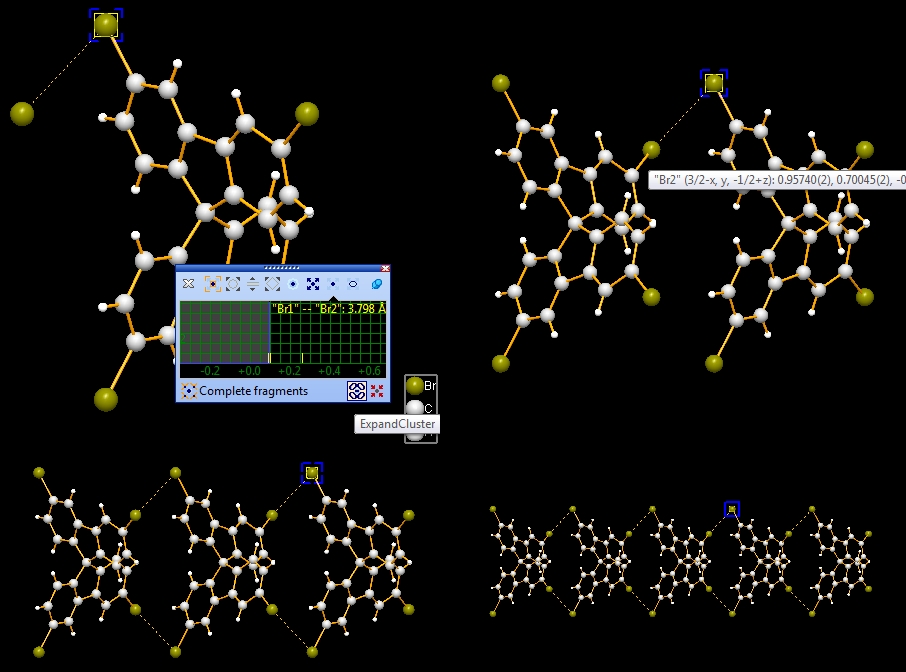
Now we make two sequences of neighbouring molecules using (a) the Br1--Br2 contact only, or (b) Br1--Br2 plus Br1--C11 contacts. To change from contact sphere evaluation (mouse wheel changes search radius) to expansion (a mouse wheel tick adds another contacted neighbouring molecule each), press the "Expand Cluster" button in the control window (see picture below).
(a)
The iteration via the Br1--Br2 contact only leads to the following sequence: (b) The iteration involving both Br1--Br2 and Br1--C11 contact leads to the following sequence: 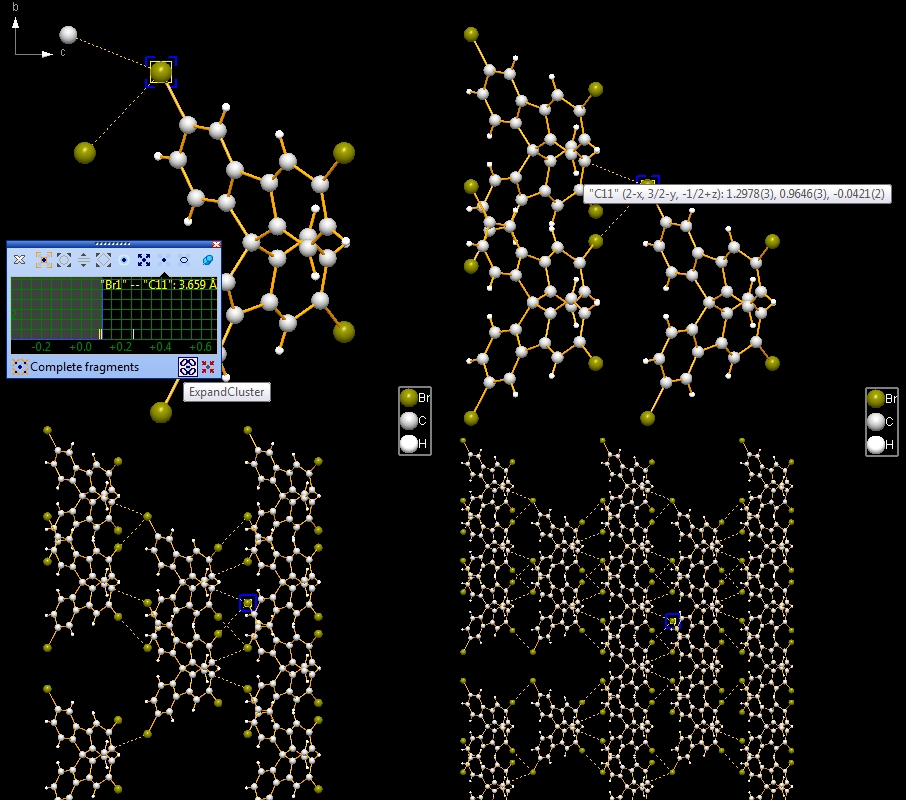
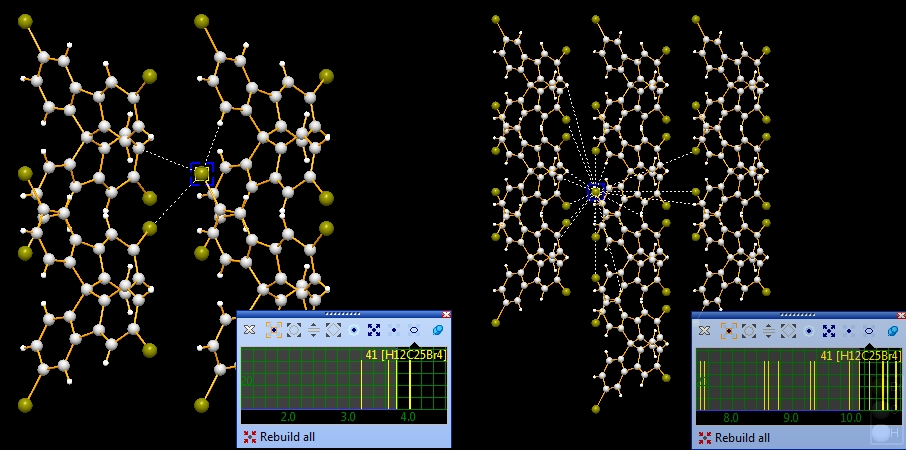
Molecule cluster
Again we start with the example COD:1501635. Best is to close and re-open the file
COD-1501635-molecule.diamdoc and start again with the single molecule.
Checking the whole molecule's neighbourhood
Previous article: Pump Up or Shrink Polymeric Frameworks Reference: COD:1501635 - Ulrich Darbost, Janie Cabana, Eric Demers, Thierry Maris, James D. Wuest: Molecular Tectonics. Use of Br...aryl Supramolecular Interactions for the construction of Organized Networks from 9,9'-spirobifluorene in the Crystalline State. CheM, 1, 52-12369 (2011). |
|
Page last modified October 05, 2023. Copyright © 2023 Crystal Impact GbR. All rights reserved. Contact Webmaster |