Diamond 4: Enhanced productivity
< Previous: Automatic and batch structure picture creation
Go to Diamond 4 Features Overview...
Next: Extended functionality for molecules and polymers >
New mode with intuitive rotation, shifting and zooming
There is a new (additional to what we now call "Selection mode") working mode, the
"Grab mode", offering you rotation or shifting of your structure picture by
grabbing an atom with the left mouse button, shifting the structure with the right mouse
button pressed, or changing the enlargement factor with the mouse wheel. Doing this,
you still have the choice to select objects. (Hold the Ctrl or Shift key when clicking
an object.). So you need not switch between the normal
selection mode and one of the different tracking modes, such as "Move/Rotation along x/y axes" etc. - although
those modes are
still available.
Neighbouring preview
You can use the mouse wheel
button to activate a (semi-transparent) preview of the atoms and bonds (molecules, polyhedra, etc.) in
the neighbourhood of the atom(s) under the mouse cursor. Use the mouse wheel to
increase or decrease the preview search sphere. Use another click on the wheel button
to take the preview into the picture.
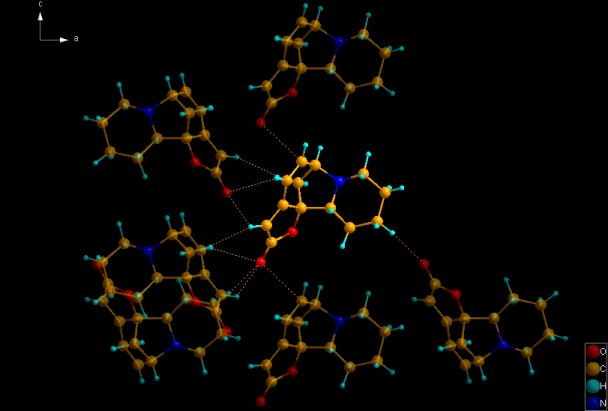
Molecule of securinine (COD:2010417) with previewed neighbouring molecules
Individual editing of properties or the surroundings using the mouse wheel
Besides the neighbouring preview mentioned above, there are several functions that
can be initiated with the mouse wheel button and settings altered with the mouse wheel:
- Change the enlargement factor.
- Zoom in/out or change perspective impression.
- Change
atom radii or thick bond tubes' radii.
- Blow up polyhedra or coordination spheres, i.e. increase or decrease oordination
number.
- Find contacts to neighbouring atoms or molecules.
- Expand from one or some molecules to a bigger cluster of neighbouring molecules.
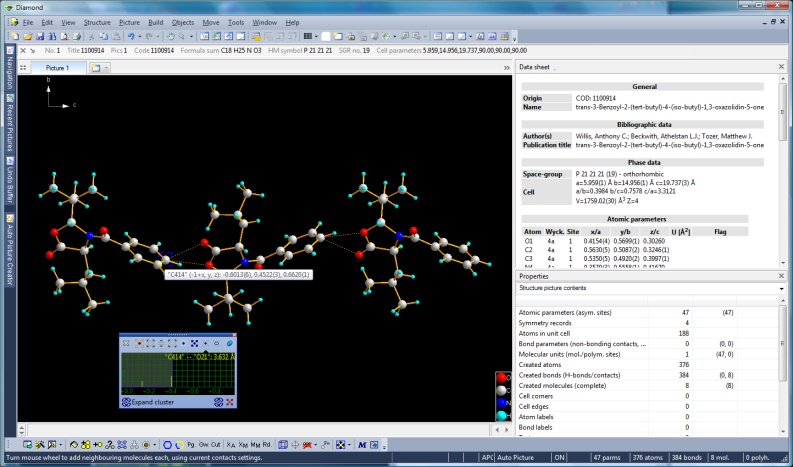
Screenshot with wheeling mode active (finding contacts) and wheeling preview window with distance histogram of contacts
(Full size image: 296 KB)
Improved evaluation of bonding spheres (connectivity)
The table of bond lengths and mean interatomic distances as well as the table of
effective radii has been improved to gain better pre-defined bonding spheres for
an imported structure. Besides this you can use the results of atom site enviroment
researches (e.g. Dirichlet domains, see below) as basis for the bonding spheres.
Define atomic environments individually to atom sites
You can define the atomic environment of every atom site individually, because
the method of bonding spheres may be too inexact and not consider different sites
of a certain atom group. The atom site environments can be derived from calculations
of their Dirichlet domains (Voronoi
polyhedra). "Atomic environment" is available
as additional option in many of the structure picture building commands that use
(coordination) spheres or search for bonds.
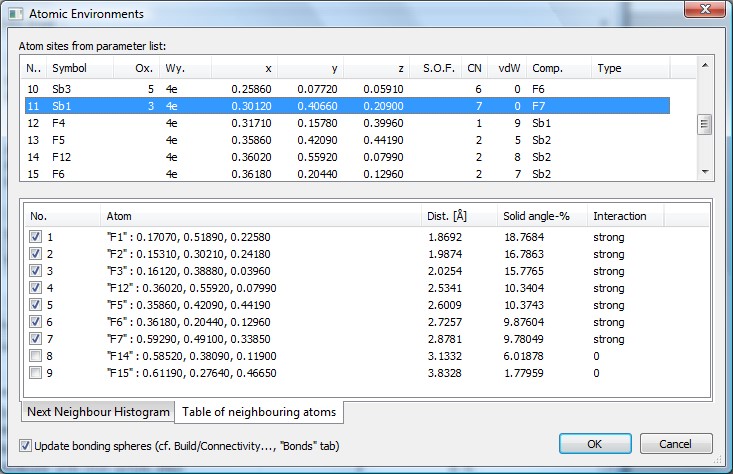
Screenshot of Atomic Environments dialog for Sb4F16 (Pearson's Crystal Data:1251073)
Quicker structure picture drawing and anti-aliasing
Structure picture drawing has been accelerated by adjusting the number of vertices
to create a big or small atom bowl etc. to the individual
situation. During rotation etc. of your structure, the vertex resolution can additionally
(and optionally) be switched down. On the other hand, anti-aliasing can be used
to reduce jagged lines.
Improved object selection
Having a structure picture with a lot of atoms and bonds as well as other objects such as labels, atom vectors
etc. it may become a difficult task to catch a certain object by clicking it in
the structure picture. Now Diamond offers a selection filter
that enables e.g. to click a vector under a bond. Besides the normal selection mode,
Diamond also offers additive and subtractive selection. So you need not hold the
Ctrl or Shift key down when you [un]select a lot of objects in the structure picture.
< Previous: Automatic and batch structure picture creation
Go to Diamond 4 Features Overview...
Next: Extended functionality for molecules and polymers >
|

