|
|
||||||
|
|
|
|
Download | |||
Diamond Version 5 User Manual: Modes of operationNeighbouring PreviewGet a preview of the atoms and molecules around the atom (molecule) under the mouse cursor
In this article:
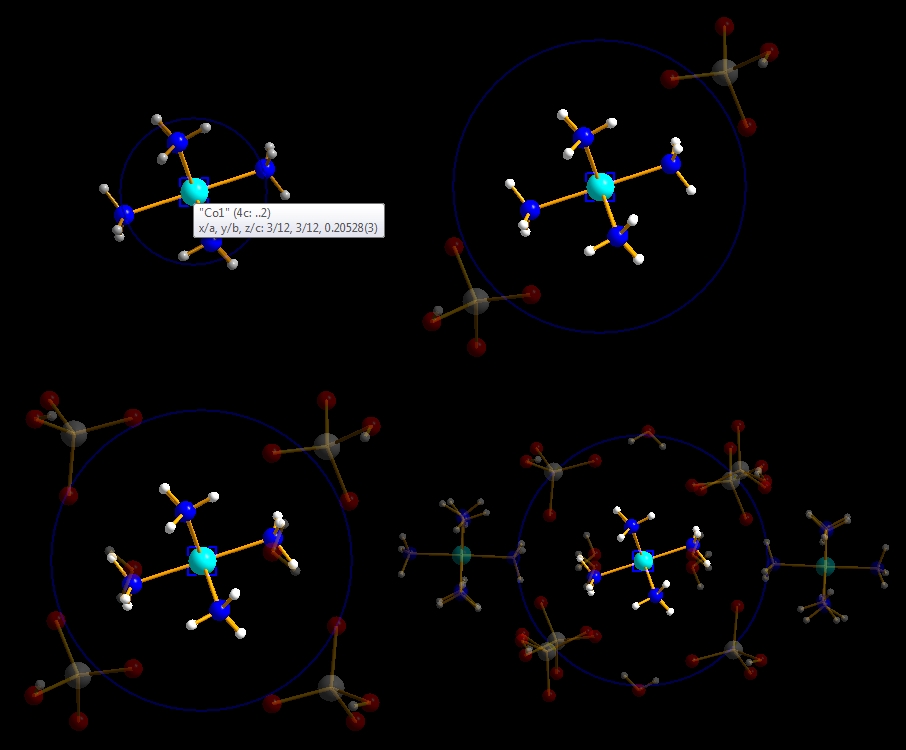
Previous article: Grab Mode To demonstrate the neighbourhood preview, we use the sample file "COD-1500005-one-Co(NH3)6-molecule.diamdoc". We also change from the default "select and edit" mode to "grab mode" by clicking on the hand symbol in the main toolbar or using accelerator key Ctrl+G or using the command "Grab Mode" from the "Edit" menu. This changes the mouse cursor to the hand symbol. If you are not yet familiar with "grab mode", please read the article "Grab Mode". Neighbourhood preview in hovering mode To start the neighbourhood preview, run the command "Neighbourhood Preview" in the "Tools" menu or simply press the F8 function key. The mouse cursor changes to a combination of hand and crosshairs.
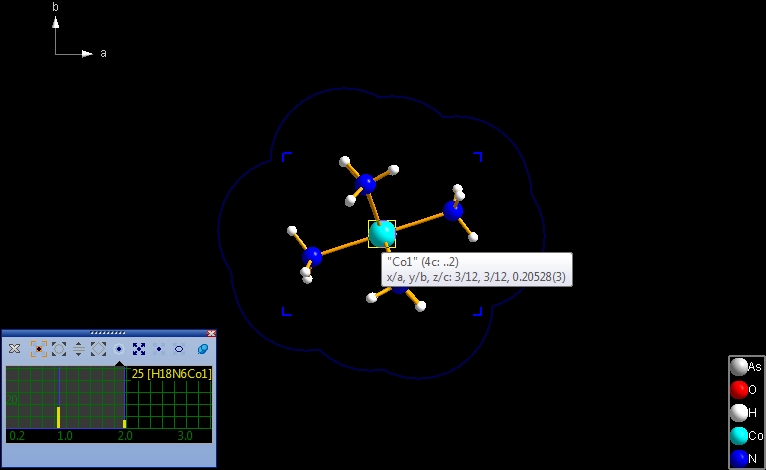
(1) Move the mouse cursor to the "Co1" atom in the center of the Co(NH3)6 molecule.
Diamond emphasizes the center of the preview with a blinking blue rectangle and
the current sphere's boundary with a blue circle. The status bar informs you that
the current size is 1.97 Angstroem, which is the size of the coordination sphere
of "Co1" with its six direct N neighbour atoms.
This kind of neighbourhood preview is called "hovering" because the semi-transparent neighbouring atoms/molecules vanish when you move the mouse cursor out of the preview sphere (i.e. leave the blue circle). When you return to the same central atom, the preview mode continues with the same sphere size, so that the same neighbours as before appear. To see the neighbourhood of another central atom, move the cursor outside the blue boundary sphere and then to the other central atom. Permanent (non-hovering) neighbourhood preview mode To prevent loosing the previewed neighbourhood when leaving the sphere boundary, click with the mouse wheel into the preview sphere's center. The mouse wheeling control window appears indicating that you have turned the hovering neighbourhood preview into non-hovering mode. This is useful to get infos about atoms lying outside the preview sphere (info tip). You can also click on other atoms than the center of the preview sphere to select it. Press the pin button in the top right to prevent the control window from closing after a few seconds. (The control window is also used for the other mouse wheeling modes -- this is described in more details in the next article: "Mouse wheeling".)
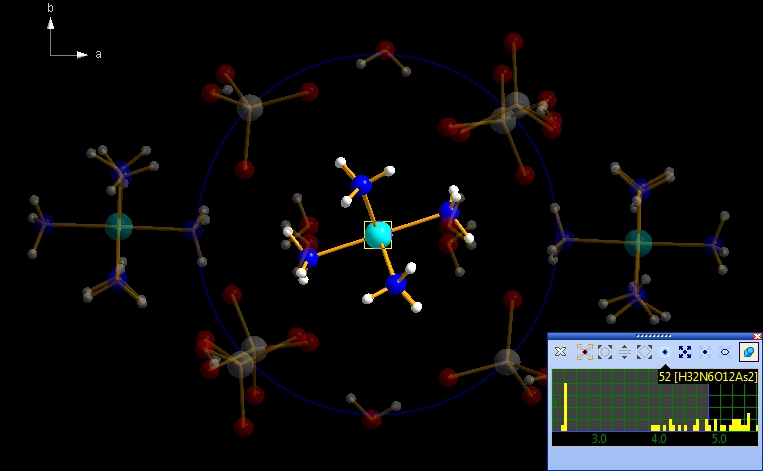
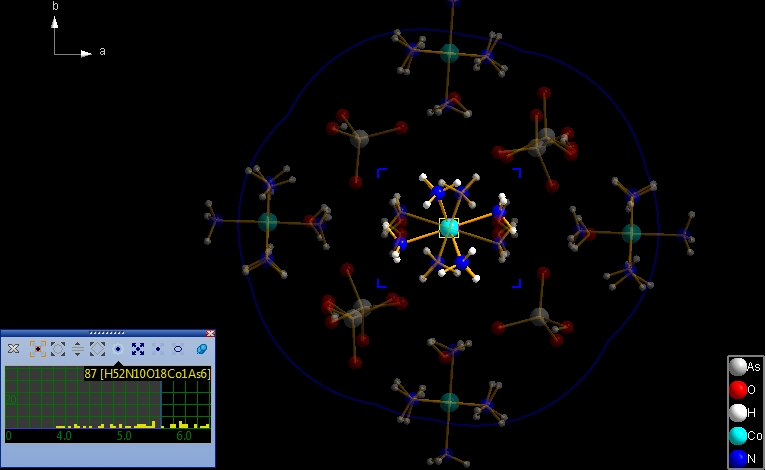
To terminate the neighbourhood preview mode, click on the 'x' symbol in the top left of the wheeling mode control window, or simply press the Escape key. The semi-transparently previewed neighbouring atoms and bonds will then disappear. Preview the neighbourhood of a whole molecule While the examples above use a single atom as the neighbourhood's center only, we now want to check the neighbourhood of an entire molecule. For that make a long click on the "Co1" atom, that means press down the mouse wheel button and hold it down for longer than one second. The animated blue rectangle enclosing the "Co1" atom explodes onto the corners of the rectangle enclosing the molecule. Instead of a single blue circle you see the outlines of the spheres of all constituent atoms of the molecule. (Note: The mouse wheel click leads us directly into permanent - non-hovering - mode.) The screenshot below shows the start of the neighbourhood preview of the Co(NH3)6 molecule...
... and after some mouse wheel ticks we come to the following scenario:
Previous article: Grab Mode Reference: COD:1500005: Hexaammine cobalt(III) hydrogenarseniate tetrahydrate - Pccn (56) - Ritu Bala, Raj Pal Sharma, Rajni Sharma, Juan M. Salas, Miguel Quiroos, William T.A. Harrison: Cationic cobaltammines as anion receptors: Synthesis, characterization an X-ray structure of bis-(hexaamminecobalt(III)) tris-(hydrogenarsenate) tetrahydrate - Journal of Molecular Structure, 828, 174-180 (2007). |
|
Page last modified December 25, 2022. Copyright © 2022 Crystal Impact GbR. All rights reserved. Contact Webmaster |