|
|
||||||
|
|
|
|
Download | |||
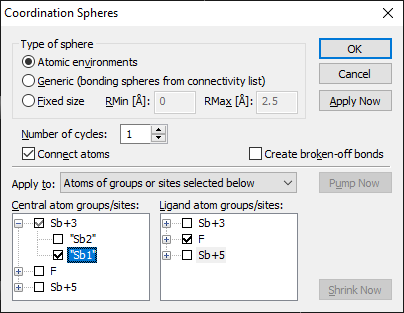
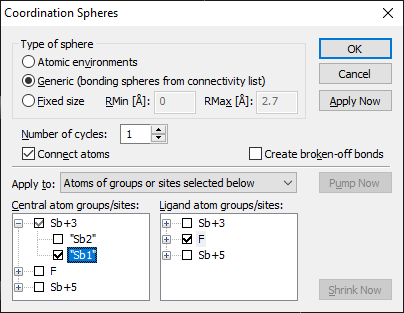
Diamond Version 5 User Manual: Building up structural partsFilling SpheresDiamond uses two different kinds of spheres, often called "enclosure spheres", to generate atoms within a spherical environment of a central atom. The simplest one is called "fixed sphere", since it is simply defined by a minimum and maximum radius value. The other one is called "generic sphere", since the size of that kind of sphere depends on the atom types (connectivity) or the atom sites (atomic environments). For details, see Fixed vs. Generic Spheres". "Filling Fixed Spheres" describes how to fill a fixed sphere around one or more selected atoms, whereas "Filling Coordination Spheres Around Selected Atom Types", "Filling Coordination Spheres Around Selected Atoms", and "Filling Coordination Spheres in Multiple Cycles" explain how generic coordination spheres can be filled. More features are available when using the "Coordination Spheres" dialog.
Previous article: Generating bonds directly Fixed vs. Generic Spheres
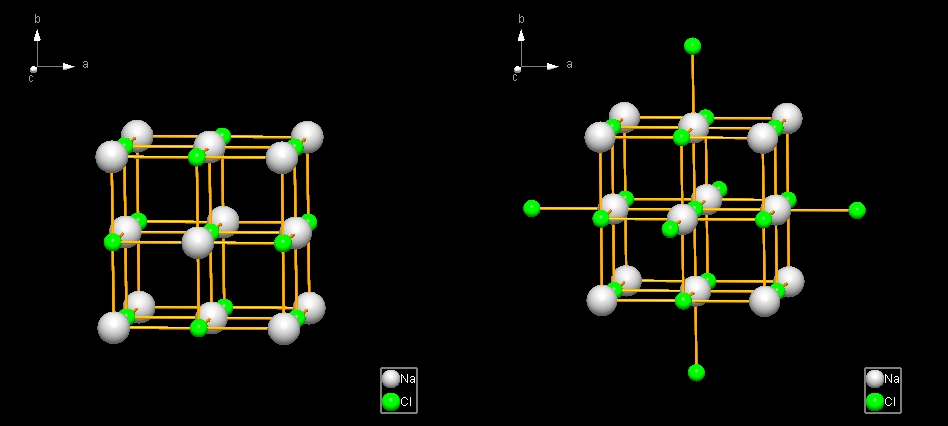
Diamond distinguishes between two kinds of spheres to generate atoms within a spherical environment of a central atom:
1. Fixed spheres
This kind of sphere is called "fixed" because its dimension is simply defined by a minimum and a maximum radius value. This means, if you fill a fixed sphere around a central atom, Diamond will create all atoms that have a distance from the central atom of at least the minimum radius value but not more than the maximum radius value. These values have the dimension Angstroems in Diamond.
2. Generic spheres
This kind of sphere is called "generic sphere" because it is used to generate neighbouring atoms which are connected with the central atom. Since these bonding spheres depend on the atom types of the atom in the center and the atom to be generated, the size of this kind of sphere varies and is not fixed. Thus "coordination spheres" are sometimes called "generic spheres" in Diamond. When filling generic spheres, a new (neighbouring) atom will only be created, if the atom type of that new atom is connected with the central atom and the distance lies within the bonding sphere range. These settings are defined in the connectivity (compare "Connectivity, part 1: Bonding spheres"). Central atom and new atom will be connected with a bond corresponding to the settings for the bond group defined for the atom group combination (compare "About atoms, atom groups, bonds, and bond groups"). Alternatively you can use an enclosure sphere that is defined on atom site rather than atom type or group level. This is defined as atomic environment and described in the article "Atomic environment". Please note that atoms may be not created, if the Filter function has been activated. To get more information about the Filter function, read the article "Filter".
"Coordination Spheres" dialog and shortcuts
One single command to handle spheres of fixed and variable size
Options of the dialog:
Shortcuts for (coordination) spheres There are five shortcuts available for this command and the functions used from the "Coordination Spheres" dialog:
The commands from the dropdown menu offer quick access to the most important functions that are available in the dialog window that is opened when you use the Coordination Spheres command from the Build menu instead:
You can find more details about "pumping" and "shrinking" of coordination spheres in the article: Pump Up or Shrink Polymeric Frameworks.
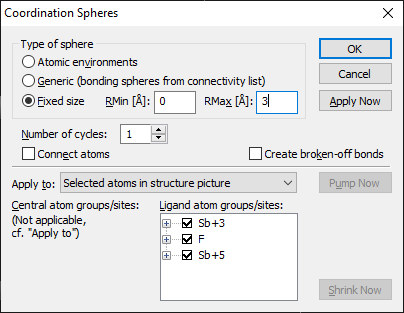
Filling Fixed SpheresUse the dialog to setup the two boundary values of the fixed shphere (RMin and RMax). The next time you run the Fill Fixed Spheres Directly command, Diamond will use these values, unless you change them in a later call to the "Coordination Spheres" dialog.
To generate atoms within fixed spheres, use the following two steps:
1. In the structure picture, select the atoms which become centers of the fixed spheres. If no atom is selected, every atom will be taken as central atom.
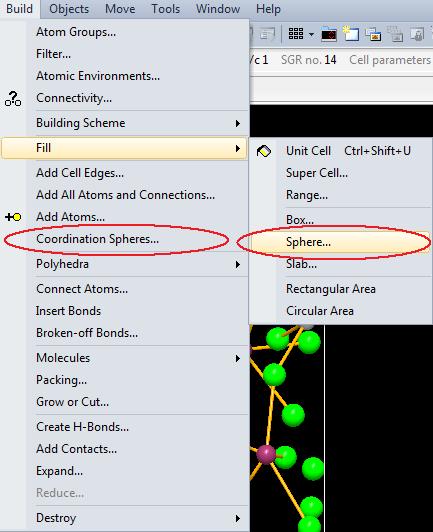
2. Run the Coordination Spheres command from the Build menu to open the Coordination Spheres dialog.
Choose Fixed size as sphere type and define the dimensions of the fixed sphere and start the generation of atoms with OK.
3. Provided you apply to selected atoms, if atoms are selected, or all atoms, if no atom is selected, and the neighbouring atoms shall not be connected with the central atom each, you can continue with the shortcut Fill Fixed Sphere Directly, since Diamond registers the latest used fixed size sphere boundaries in its Windows registry settings. If you need different options, you have to use the dialog again. (Here you can use the Apply Now button to repeatedly fill (fixed size) spheres.)
"(Fixed) sphere" also available in the "Fill" sub-menu
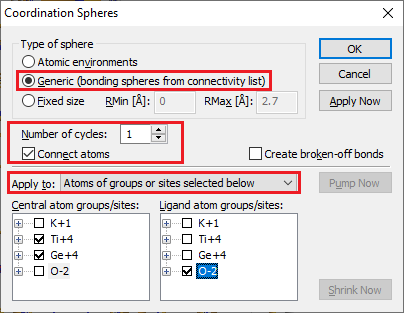
Filling Coordination Spheres Around Selected Atom GroupsThe generation of atoms within coordination spheres can be performed in different ways. This part of this article describes how coordination spheres around central atoms belonging to selected atom groups (atom types) are filled with atoms belonging to other selected atom groups. To fill coordination spheres around selected atom groups, choose the Coordination Spheres command from the Build menu. This will open the Coordination Spheres dialog:
Ensure that Apply to is set to Atoms of groups or sites selected below is activated. Choose the atom group(s) which shall become centers of coordination spheres in the Central atom list, and the atom group(s) of the atoms to be generated in the Ligand atom list. Leave the Number of cycles input field 1, unless you want to fill coordination spheres in multiple cycles (compare "Filling Coordination Spheres in Multiple Cycles"), and do not check the checkbox Create broken-off bonds, unless you want to create "broken-off" bonds (compare "Creating and modifying broken-off bonds"). To start the generation of atoms within the coordination spheres, either press the OK button, which will close the dialog, or press the Apply Now button, which does not close the dialog and has the advantage that you can repeat the procedure with the same or other atom groups.
A new atom will only be created in this procedure, if:
(1) its atom group belongs to the atom types selected in the Ligand atom list,
(2) the atom group of the central atom belongs to the atom groups selected in the Central atom list, (3) the atom group combination (bond group) is enabled for being connected with a bond in the connectivity list, (4) the distance between new atom and central atom lies within the range of the bonding sphere, which is also defined in the connectivity list, (5) the new atom is not blocked by the Filter function. If atoms have been selected in the structure picture, that selection will be ignored within this procedure.
Filling Coordination Spheres Around Selected Atoms
While the previously described procedure (compare "Filling Coordination Spheres Around Selected Atom Groups")
ignores the current selection of atoms in the structure picture but takes all atoms belonging to selected atom types as central atoms,
this part describes how to take the currently selected atoms as centers of coordination spheres only.
To fill coordination spheres around selected central atoms, first select the atoms. If no atom is selected, every atom will be taken as central atom.
To start the generation of atoms, either press the (red marked) button
A new atom will only be created in this procedure, if: (1) the atom group combination (bond group) of central atom and new atom is enabled for being connected with a bond in the connectivity list, (2) the distance between new atom and central atom lies within the range of the bonding sphere, which is also defined in the connectivity list, (3) the new atom is not blocked by the Filter function.
Please note:
Filling Coordination Spheres in Multiple Cycles
A common procedure is the stepwise construction of coordination spheres especially in 3D frameworks or layers of inorganic structures. That means you first create atoms within the coordination spheres of selected central atoms and then repeat this procedure for the just created atoms and so on.
The stepwise filling of coordination spheres can be done in three different ways:
1. By clicking on the 2. By choosing the Coordination Spheres command from the Build menu and using the Apply Now button in the "Coordination Spheres" dialog.
3. By using a number greater than one in the input field Number of cycles in the "Coordination Spheres" dialog.
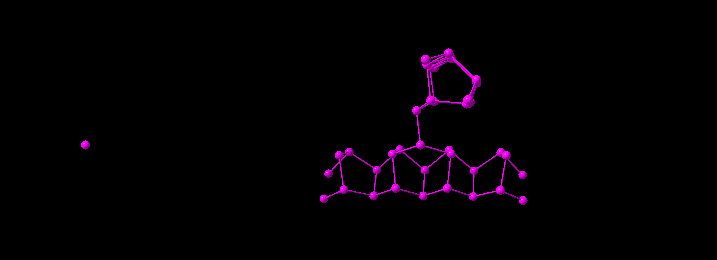
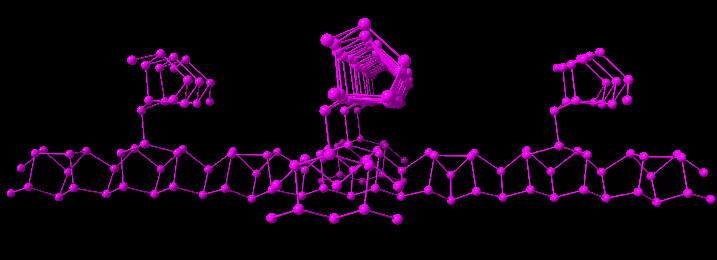
Example: Building up the 3D framework of Hittorf violet phosphorus, starting with one atom of the parameter list (upper left), and then calling the coordination sphere function five times, leading to the picture right above. Applying the coordination sphere function again with a number of cycles of ten leads to the picture below.
If you build up coordination spheres stepwise without using the "Coordination Spheres" dialog (method #1), you should ensure that no atom is selected. In the first step the coordination spheres of all atoms will be filled, while in the second step all coordination spheres of the atoms that have been created in the first step will be created, and so on. If you use method #3, Diamond swaps the selection of atom groups for central atoms and ligand atoms after each step. For example, if you have selected "Si" as central atom and "O" as ligand atom, and a cycle number of 3, in the first cycle all O atoms in the coordination spheres of all Si atoms will be created, while in the second cycle all Si atoms will be created in the coordination spheres of all O atoms. The third cycle will work like the first cycle, that means Si as central atom.
Features of the "Coordination Spheres" dialog
Access to individual atom sites
Multiple cycles now works through already complete spheres
"Pump up" and "shrink"
Atomic environment as new sphere type option
Connection between center and ligands now independent from sphere type
Previous article: Generating bonds directly
Reference for PCD-1251073: |
|
Page last modified July 22, 2022. Copyright © 2022 Crystal Impact GbR. All rights reserved. Contact Webmaster |
 or the hotkey Shift+Ctrl+S, you can fill coordination spheres around the selected atoms (or around all atoms,
if no atom is selected). There will be just one cycle, using the bonding ranges from the connectivity or the atomic environments,
depending on the latest settings defined in the "Coordination Spheres" dialog. (Default is "generic spheres" from connectivity.) Any selection concerning
central and ligand atom groups that may be done previously in the dialog that
is opened through the menu command, are ignored here.
or the hotkey Shift+Ctrl+S, you can fill coordination spheres around the selected atoms (or around all atoms,
if no atom is selected). There will be just one cycle, using the bonding ranges from the connectivity or the atomic environments,
depending on the latest settings defined in the "Coordination Spheres" dialog. (Default is "generic spheres" from connectivity.) Any selection concerning
central and ligand atom groups that may be done previously in the dialog that
is opened through the menu command, are ignored here.