|
|
||||||
|
|
|
|
Download | |||
Diamond Version 5 User Manual: Building up structural partsDirichlet DomainsDetermination of atom site environments basing upon Dirichlet domains (Voronoi polyhedra)
This article is about:
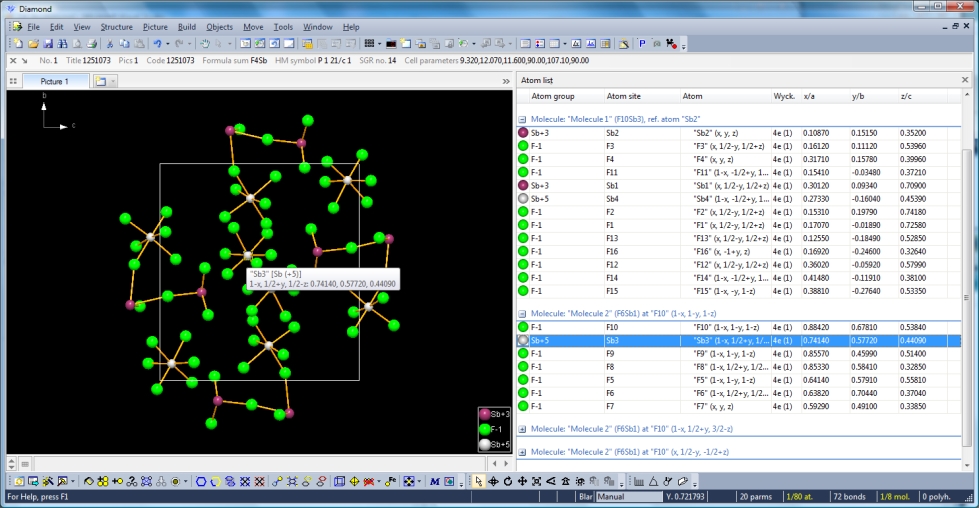
Previous article: "Atomic environment" as additional (optional) criterion when filling a coordination sphere or adding coordination polyhedra Open the sample file "PCD-1251073.diamdoc". This is a 1:1 adduct of antimony trifluoride and antimony pentafluoride containing two types of antimony, Sb+3 and Sb+5, in the atomic parameter list. The picture uses the bonding spheres derived from ionic radii, and the picture shows a packing diagram with two types of molecules: (To activate the atom list, run "Atom List" from the "View" menu, and in the atom list run "Arrange by molecules" in the context menu of the atom list.) Please note: The screenshots have been made with Diamond version 4, the functionality is the same in the current version 5.
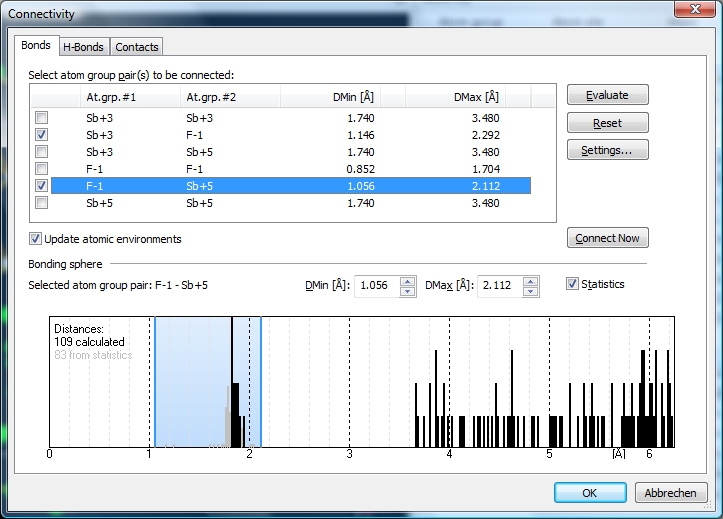
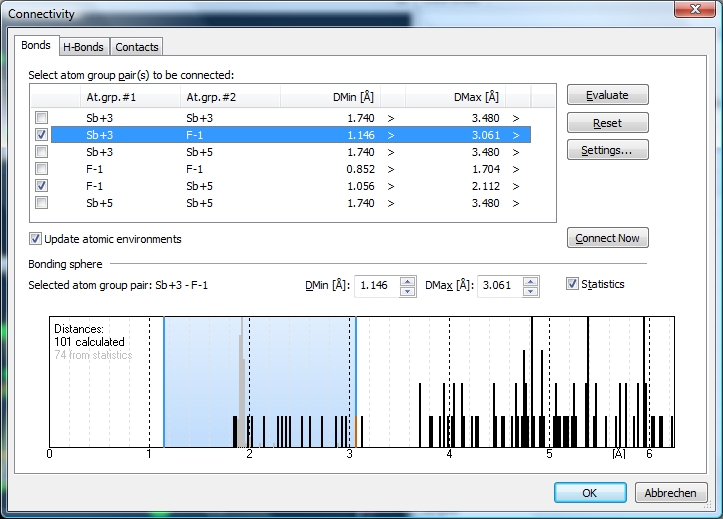
There are four Sb atoms in the atomic parameter list, two of them Sb+5 ("Sb3" and "Sb4"), and Sb+3 the two others ("Sb1" and "Sb2"). Let us check the bonding spheres on the "Bonds" page of the "Connectivity" dialog ("Build" menu). The distances histogram for Sb+5--F-1 has a sharp range of bonding distances and a wide and significant gap to the next spheres' distances ...
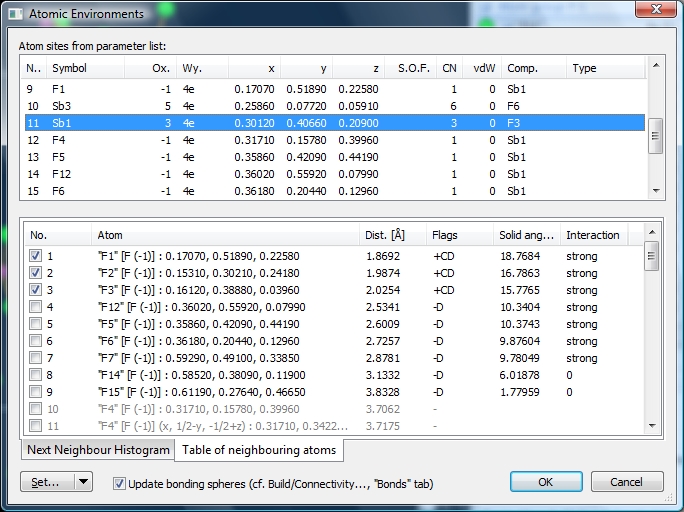
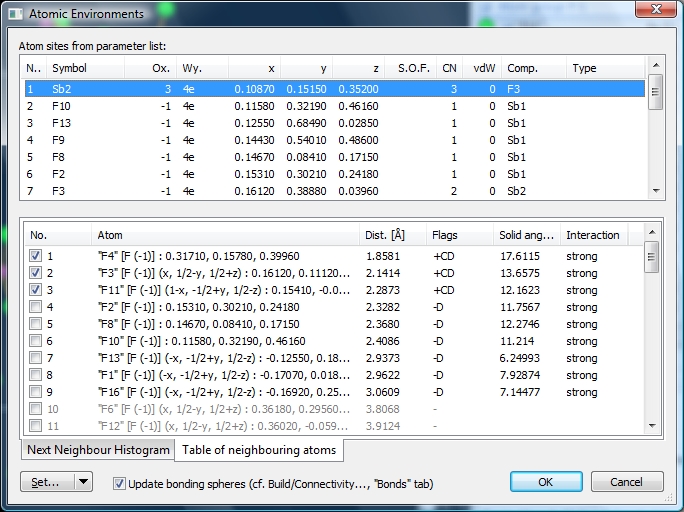
... whereas the histogram for the Sb+3--F-1 distances is not significant, and the upper bonding sphere boundary falls arbitrarily into a scattered sequence of Sb+3--F-1 distances. There is a gap at approximately 3.2 Angstroms. We also cross-check the atomic environments ("Build" -> "Atomic Environments"). When we mark the "Sb1" site in the upper list, the lower list shows the atoms in the environment. Atoms #1 through #9 are in black, and there are more in grey at positions #10 etc. Atoms #1 through #9 ("F1", ..., "F15") belong to the Dirichlet domain of the site "Sb1" (flag 'D'), the first three of them have been captured by the bonding sphere generation function (cf. "Connectivity") (flag 'C') and are thus marked as bonded (flag '+'). The remaining six F-1 atoms follow after a distance gap but only the first four of them have "strong" interaction. Some of the subsequent atoms ("F4") do not more belong to the Dirichlet domain, although they are closer to the center than "F15".
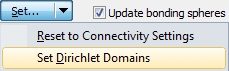
Now looking at site "Sb2" we have three F-1 neighbours marked as bonded but - as you see - the upper sphere boundary falls arbitrarily into a scattered range of Sb--F distances. There are gaps between neighbour #6 and #7 as well as a significant gap beyond the Dirichlet domain, that means after atom #9 ("F16"). All nine atoms of the Dirichlet domain show strong interaction.
We now want to take the strongly interacting atoms from the Dirichlet domains and overwrite the bonding sphere boundaries that have originally been generated simply from effective (ionic) radii. We use the command "Set Dirichlet Domains", available from the menu activated by the dropdown arrow right beneath the "Set..." button:
After application of the command "Set Dirichlet Domains", site "Sb2" has nine bonded F-1 neighbours, whereas site "Sb1" has seven:
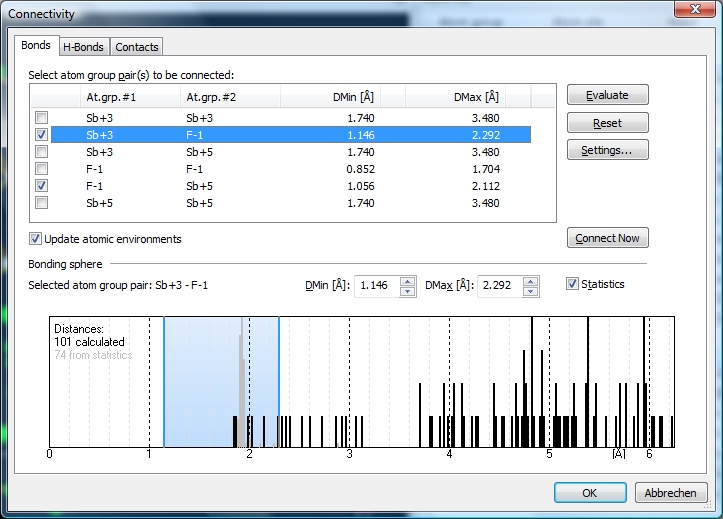
Before you close the dialog with "OK", assure that the checkmark at "Update bonding spheres [...]" is set. (This will update the bonding spheres. This checkmark is set by default. If you want atomic environments deviating from the bonding spheres, for instance when building special coordination polyhedra, you should clear this checkmark.) You may - again - cross-check the "Connectivity" dialog. The distance histogram for Sb+3--F-1 now looks like this. The single distance before the gap that does not belong to the bonding sphere, comes from "F14" that belongs to the Dirichlet domain of "Sb1" but is not strongly interacting.
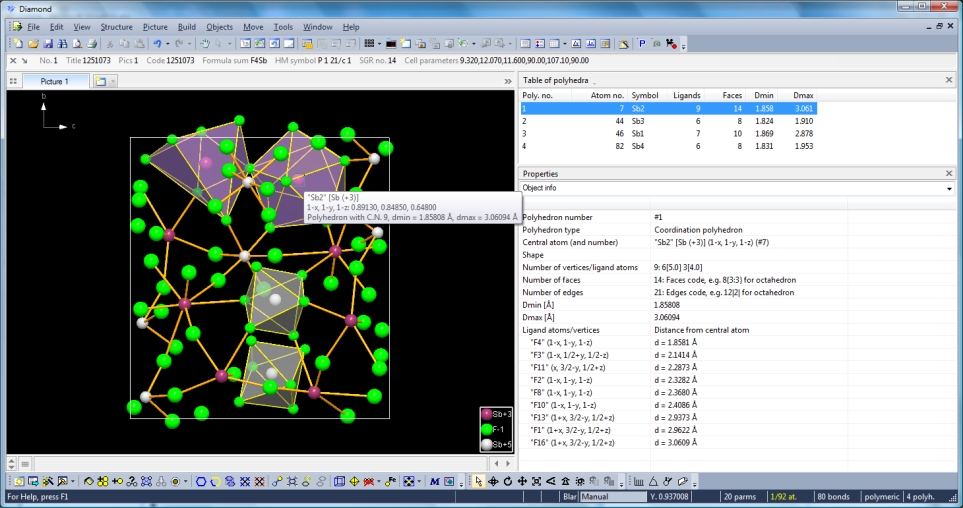
If you now destroy the previous picture ("Build" -> "Destroy" -> "All"), run "Build" -> "Fill" -> "Unit Cell" and build one coordination polyhedron around the four Sb sites each, you get something like in the screenshot below. (Polyhedron designs may deviate depending on your current settings in Diamond. Here we use semi-translucent faces in violet for the polyhedra around Sb+3, and light gray ones around Sb+5. The table of polyhedra in the data pane is available through: "View" -> "Table" -> "Polyhedra". This picture has been saved in the sample document "PCD-1251073-unit-cell-with-Sb-polyhedra.diamdoc".)
This article is closely correlated with the article "Voronoi Polyhedra". You should check that article to see the Voronoi polyhedra. (Note: In Diamond, we use the name "Dirichlet domain" for the atomic environmental description, whereas "Voronoi polyhedron" is the 3D graphical object, similar to the coordination polyhedra, but with intersections between planes as vertices rather than real atoms.)
Previous article: "Atomic environment" as additional (optional) criterion when filling a coordination sphere or adding coordination polyhedra
Reference: |
|
Page last modified January 24, 2023. Copyright © 2023 Crystal Impact GbR. All rights reserved. Contact Webmaster |