|
|
||||||
|
|
|
|
Download | |||
Diamond Version 5 User Manual: Building up structural partsAdding and Finding Symmetry-equivalent Molecules
In this article:
Previous article: Completing molecular fragments Add Molecules dialogTo prepare the next structure picture we start from sample "COD-1501635-unit-cell-packings.diamdoc", which has already four pictures of packings, and create a fifth picture ("Picture 5") as blank picture with current settings (that means the same atom designs, viewing direction, etc.). You may call the "New Picture" command from the "Picture" menu and walk through the "New Structure Picture Assistant", but it is easier to click on the main toolbar's button New Blank Picture or to use the accelerator key Alt+Ctrl+N:
If you are using tabs, you can also click on the dropdown button right beneath the "Create new tab" pseudo-tab and choose the command "New Blank Picture With Current Settings" from the dropdown menu:
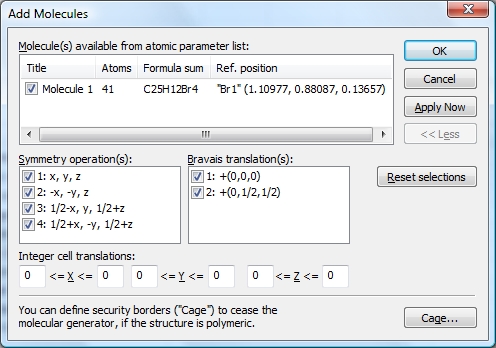
The Add Molecules command is available in the Molecules sub-menu of the Build menu and opens the Add Molecules dialog, which looks and behaves similar to the Add Atoms dialog that creates selected atoms of the parameter list directly at "x,y,z" or at symmetry-equivalent positions. Here you have a list of the molecular units rather than the list of atom sites.
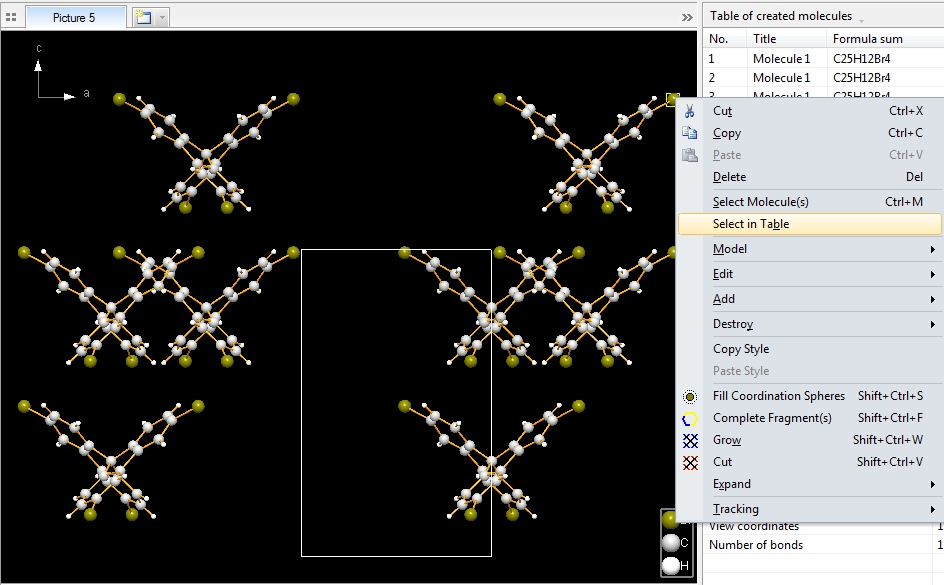
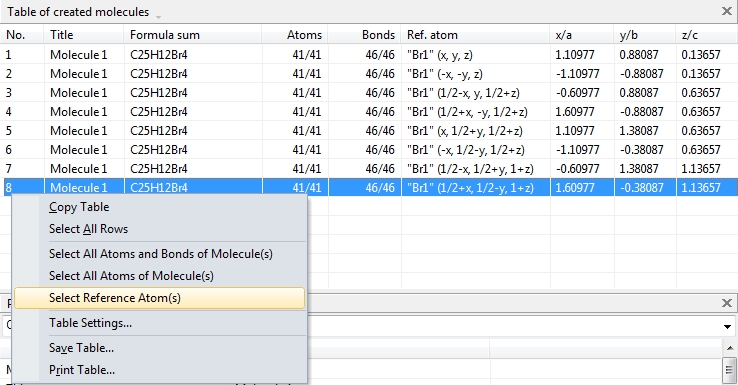
To create the picture in the following screenshot, we mark all symmetry operations and Bravais translations of space group Aba2 (no. 41), so we should get eight molecules. In this compound this is rather straightforward, since all atoms in the parameter list are on the general site 8b, except the spiro atom "C1" on 4a (0, 0, z). The data pane shows the table of created molecules (cf. "View"/"Table"/"Created Molecules"), to get to know what molecule comes from what symmetry operation. To select the molecule row in the table, click on an atom of a molecule in the structure picture and choose "Select in Table" from the context menu.
This also works the way back. Select a row in the table of created molecules and choose the command "Select Reference Atom". The two neighbouring commands "Select All Atoms of Molecule" and "Select All Atoms and Bonds of Molecule" select all atoms (and bonds, rsp.) of the molecule that is highlighted in the table of created molecules. (This also works for multiple highlighted rows and selects multiple molecules.)
If you want instead to find neighbouring or closest molecules to your molecule of interest in a structure picture, it is better to use the "Find Molecules" dialog or to make use of the "expand" functionality in Diamond, which will be described in the article "Expand or reduce clusters of molecules", rather than trying to get the wanted molecules by trial-and-error of symmetry operations and/or integer cell translations in the "Add Molecules" dialog. Find Molecules dialogHere you find a list of the molecules around the currently selected atom or the selected molecule within a sphere of given size. (It does not consider vagabonding atoms nor atoms belonging to polymers.) You can destroy a selected molecule or run the "Complete fragment" command, if the selected molecule is not yet complete, i.e. typically cut during a building command that uses a box etc.
Previous article: Completing molecular fragments
References: |
|
Page last modified August 12, 2022. Copyright © 2022 Crystal Impact GbR. All rights reserved. Contact Webmaster |